Long-Read Sequencing and Electronic Genome Mapping
Long-range genomic information is often missed with short-read sequencing. However, this information is crucial for detecting large-scale structural variants, identifying alternative gene splicing isoforms, or assembling entire genomes (see more applications below). In such scenarios, long-read sequencing is indispensable. At NUSeq, we offer long-read sequencing services using both Oxford Nanopore and PacBio Revio platforms. The major applications of long-read sequencing include:
- Structural variant (SV) detection
- Single nucleotide variant phasing
- Full-length transcript sequencing and splicing isoform detection
- Detection of fusion transcripts
- De novo genome assembly
- Direct detection of epigenetic base modifications on DNA and RNA
For structural variant detection, the core is also equipped with a new technology called Electronic Genome Mapping (EGM). This technology detects SVs greater than 100 kb and as small as 300 bp, complementing long-read sequencing as well as cytogenetic techniques.
Long-Read Sequencing Platforms
Electronic Genome Mapping
The OhmX system from Nabsys powers EGM at NUSeq. Using solid-state nanochannels, this platform electronically detects tagged high-molecular-weight (HMW) DNA. Features include:
- Resolution: Detection of structural variants from 300 bp to >100 kb
- Labeling: High-density tags at known recognition sites
- Technology: Electrical resistance changes during DNA passage through nanochannels
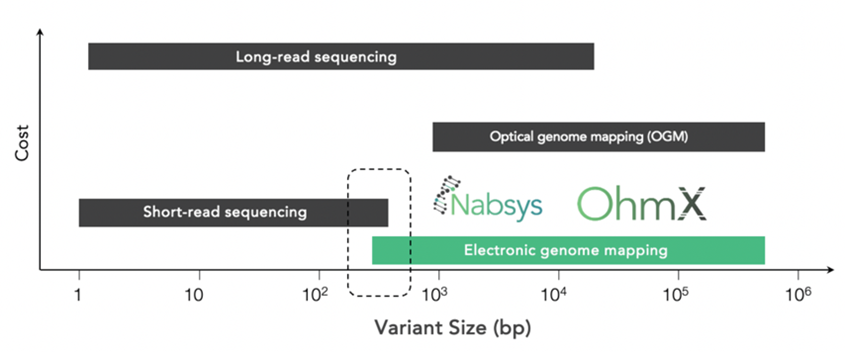
EGM is complementary to long-read sequencing in detecting structural variants. On the lower end, it also overlaps with short-read sequencing in detecting smaller SVs.
Pricing
PacBio Revio
Check the core’s Pricing page for costs associated with:
- HiFi sequencing
- SMRTbell library prep for WGS and other DNA applications
- Iso-Seq library prep for full-length RNA sequencing to detect splicing isoforms
- Kinnex library prep for full-length RNA, single-cell RNA, and 16S rRNA sequencing
Oxford Nanopore
Check the core’s Pricing page for costs associated with:
- Nanopore sequencing on MinION and PromethION flow cells
- Oxford Nanopore library prep
Nabsys OhmX
This is a recently installed technology. Please contact the core to get an estimate.
Service Request
Project consultation is provided free-of-charge. Long-read sequencing services can be requested through NUcore.
Sample Submission
For successful long-read sequencing and electronic genome mapping, HMW DNA of sufficient quality and quantity is essential.
DNA Sample Preparation:
- HMW DNA can be extracted using kits such as Nanobind and NEB Monarch HMW DNA extraction kits.
- Purity is assessed using Nanodrop, and molecular weight is determined with pulsed-field electrophoresis (NUSeq uses the Agilent Femto Pulse System).
- Accurate concentration measurement is performed using fluorometry methods, such as Qubit.
RNA Sample Preparation:
- For full-length transcript sequencing, high-quality RNA is crucial.
- RNA should be DNA-free (DNase treatment recommended).
- RNA Integrity Number (RIN) ≥ 7 and at least 500 ng of RNA are required to start library prep.
Sample Requirements:
- Whole Genome Sequencing on PacBio Revio: Library prep can begin with 500 ng of genomic DNA using the latest SPRQ chemistry. At least 70% of DNA fragments should exceed 10 kb in size (GQN ≥ 7.0).
- Electronic Genome Mapping on Nabsys OhmX: At least 5 micrograms of DNA is needed. Per Nabsys, input DNA can be prepared from 1 million fresh or frozen cells, or 500 microliters of blood, using NEB’s Monarch HMW DNA Extraction Kit for Cells & Blood (#T3050S).
- For other applications, as required input DNA/RNA amount varies from application to application, please contact the Core team for specific requirements for your project.
Bioinformatics
Data analysis is provided upon request.
Frequently Asked Questions
What are key factors in extracting HMW DNA?
To minimize degradation, use fresh or properly preserved samples. Adhere to kit instructions and use gentle cell lysis conditions. Avoid pipetting and vortexing to prevent physical shearing.
How is HMW DNA fragment size measured?
NUSeq employs the Agilent Femto Pulse System for precise measurement of DNA fragments up to 165 kb using pulsed-field electrophoresis.
What should I expect with low-quality DNA samples?
Low-quality DNA samples often contain fragments smaller than 10 kb, resulting in reduced data yield. It is not recommended to use such samples.
How does the PacBio Revio long-read sequencer work?
The Revio system is built on PacBio’s Single Molecule Real-Time (or SMRT) sequencing technology. In a nutshell, the SMRT sequencing process records signals emitted from the incorporation of nucleotides during synthesis of a complementary strand based on sequence on the template strand. Such signals, recorded into a movie format, are then used for base calling. The signal capture is achieved with the use of zero-mode waveguides (ZMWs), or nm-scale wells where single DNA molecule sequencing takes place. To improve sequencing accuracy, the system uses a process called Circular Consensus Sequencing (or CCS), which sequences the same DNA target multiple times to produce consensus high-fidelity reads (or HiFi reads).
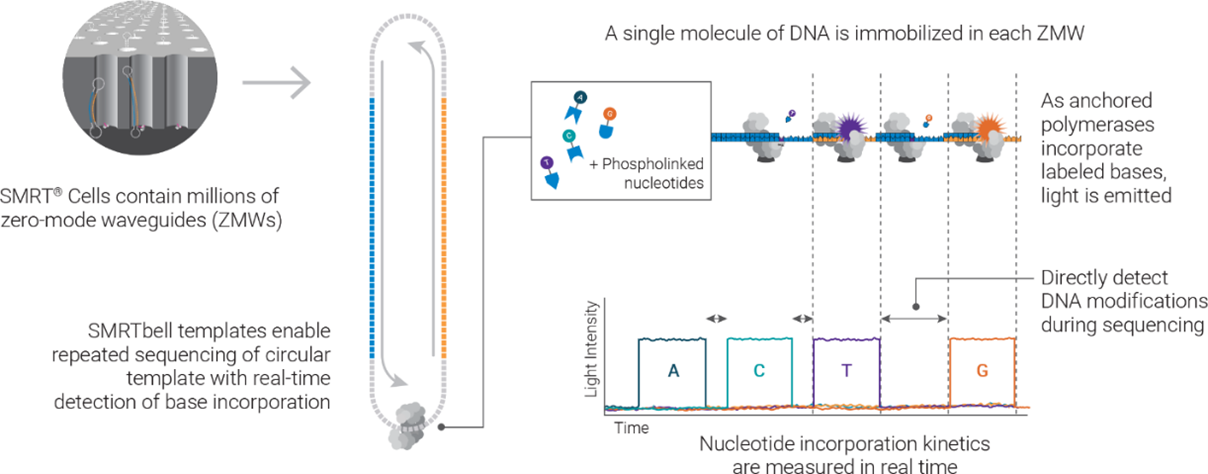
An illustration of how SMRT sequencing works. The Revio SMRT Cell contains 25 million zero-mode waveguides (ZMWs) and can generate 90 gigabases (Gb) of data per run. Using the latest SPRQ chemistry, it can produce up to 120 Gb per flow cell, enabling sequencing of two human genomes at 20x coverage. (Source: PacBio)
How does Electronic Genome Mapping (EGM) work on the Nabsys OhmX System?
OhmX employs solid-state nanochannels to electronically detect tagged HMW DNA. DNA molecules are tagged at known recognition sites and moved through nanochannels where changes in electrical resistance indicate presence of the tags. The OhmX’s onboard field-programmable gate array (FPGA) conducts real-time processing of the changes in electrical signals to determine the location of the tags. The signal-processed files are then used for SV analysis. Technical details of this technology are published in BioRxiv.
What software is used for EGM-based SV analysis?
Signal-processed files are uploaded to the cloud for SV analysis using the Human Chromosome Explorer, a software for de novo SV discovery and verification. This software is developed by Hitachi High-Tech in collaboration with Nabsys. This platform enables the detection of balanced and unbalanced SVs across a broad length scale from 300 bp to >100 kb.
How does Electronic Genome Mapping (EGM) compare to Optical Genome Mapping (OGM)?
OGM is based on optical detection of fluorescent labels. Light diffraction limits detection resolution and makes it difficult to detect labels in close proximity. Electronic detection used in EGM enables detection of labels in close proximity. The increased label density leads to greater resolution, allowing detection of smaller SVs in the range of 300-1000 bp.