ATAC-Seq for Open Chromatin Profiling
The eukaryotic genome is highly packaged to fit into the very limited nuclear space. As a result, access to genomic information is tightly regulated based on cellular state. What regions of the genome are accessible reveals a great deal about the state of the cell. ATAC-seq, or Assay for Transposase-Accessible Chromatin coupled with next-gen sequencing, is a technique to locate accessible chromatin regions.
As the name suggests, ATAC-seq is based on the use of an engineered, hyperactive transposase (called Tn5), which fragments DNA in open regions of the chromatin. In the same process, it simultaneously tags the ends of the fragmented DNA with sequencing adapters. This tagmentation process is a key part of ATAC-seq library construction.
Standard ATAC-seq: For input material, NUSeq needs 50,000 live cells to start library prep. Extra cells are needed for cell viability and density checks before conduct of library prep. Please submit at least 60,000 cells to the Core. Since cells need to be processed immediately after delivery to the Core, the user needs to contact the Core ahead of time (at least by one week) to schedule the work.
Single cell ATAC-seq using 10x Chromium: Single nuclei suspension prepared from fresh, cryopreserved, and flash frozen tissue or cell samples is needed for library prep. As performed for single cell RNA-seq, single nuclei prep will be check first for quality and concentration, and 500-10,000 nuclei can be targeted in each sample. As single nuclei prep needs to be processed right away by Core personnel upon arrival, the user needs to schedule the work ahead of time (at least by one week).
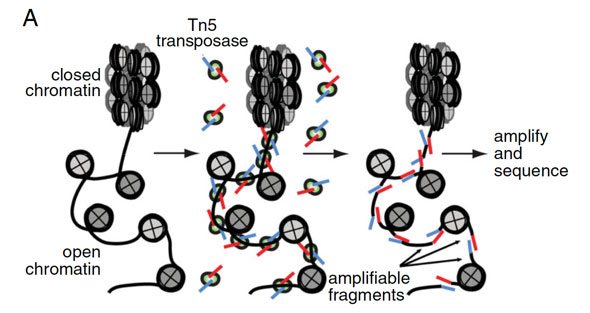
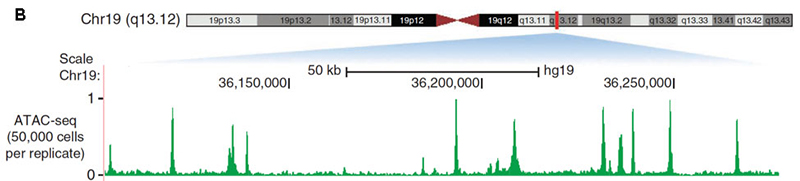
A. ATAC-seq basic scheme; B. An example of ATAC-seq signal. From Nature Methods 10, 1213–1218 (2013).
ATAC-Seq
ATAC-Seq Library Prep Pricing
Costs for standard and single-cell ATAC-seq library construction are $350 and $2,150, respectively, for Northwestern users. Please check the Core Pricing page for most up-to-date rates, including those for external and industrial users.
Service Request
Project consultation is provided free-of-charge. ATAC-seq services can be requested through NUcore. Please finish and submit our project intake form when requesting this service.
Sample Submission
Standard ATAC-seq: Live cells, not genomic DNA, is required as input for ATAC-seq. The number of cells is critical to project success, with 50,000 cells as a good starting point. Healthy cells in a homogeneous single-cell suspension work the best. Before proceeding to library prep, NUSeq staff will check for cell viability and density.
Single cell ATAC-seq: Single nuclei suspension prepared from fresh, cryopreserved, and flash frozen tissue or cell samples is needed for library prep. The 10x Genomics scATAC-seq Support page lists Demonstrated Protocols for sample prep. NUSeq staff will check single nuclei prep quality and concentration upon sample delivery.
It is essential to coordinate with core staff for sample delivery because samples need to be processed right away. It is advisable to schedule the work with the Core as early as possible, prior to cell preparation.
Sequencing Mode
Standard ATAC-seq: Single-end 50 or 75bp reads are usually enough for mapping ATAC-seq reads to the reference genome. For some applications such as nucleosome mapping, paired end sequencing (PE37, PE50, or PE75) is preferred.
Sequencing depth: at least 50 million reads per sample are recommended.
Bioinformatics
Data analysis is provided upon request. Raw data and analysis results are usually returned to user via OneDrive.
Frequently Asked Questions
What are the consequences of using inappropriate numbers of cells for standard ATAC-seq?
Using too few cells will lead to over-digestion and increased background noise, while using too many cells will lead to under-digestion and production of large fragments that are hard to sequence on Illumina sequencers.
Why do we have many mitochondrial reads in our data?
It is normal to have a fraction of reads derived from mitochondrial DNA because of their abundance in each cell.
Can I integrate my ATAC-seq data with ChIP-seq and RNA-seq data?
An integrated approach will make the best use of the data. Here is a good example of integrating bulk ATAC-seq with ChIP-seq and RNA-seq. At single cell level, it is also achievable to perform integrated scATAC-seq and scRNA-seq data analysis based on this 10x Genomics product sheet.