Microbiome Sequencing
Next-gen sequencing offers a powerful approach to study microbial diversity and dynamics in our body or other environments. For microbiome sequencing, there are two general approaches: targeted amplicon sequencing and shotgun metagenomic/metatranscriptomic sequencing. The first approach focuses on the use of hypervariable regions that are bounded by conserved DNA sequences, most typically 16S rRNA for bacteria and ITS (Internal Transcribed Spacer) rRNA for fungi. The conserved DNA sequences here are used as PCR primer binding sites to amplify the hypervariable regions, which are then sequenced and analyzed for microbial composition. The shotgun sequencing approach, on the other hand, provides random sampling of all genomes (or transcriptomes) contained in a microbiome. To carry it out, total DNA (or cDNA made from RNA) extracted from a sample is first randomly fragmented into short pieces for sequencing library construction prior to sequencing and analysis.
The shotgun approach takes an unbiased path to providing a comprehensive assessment of genome content and taxonomic composition, or functional state and diversity if studying the metatranscriptome, in a microbial community. The targeted amplicon approach provides an alternative through generating an approximate estimation of relative taxonomic abundance in the community, with the benefits of reduced data complexity and project cost. NUSeq provides full support for both approaches. For the targeted amplicon approach, the Core provides sequencing that covers the entire 16S rRNA gene in six amplicons (V1V2, V2V3, V3V4, V4V5, V5V7, and V7V9), plus the fungal ITS region. The use of all the variable regions in the rRNA gene provides more robust bacterial profiling than the use of individual regions. The sequencing of the fungal ITS region is provided at no additional cost.
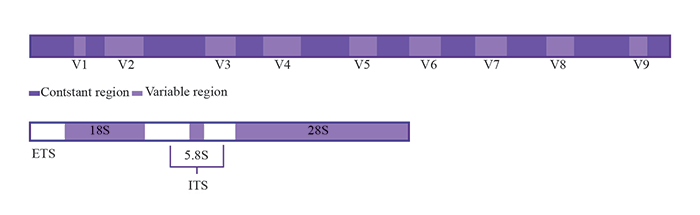
Top: Structure of the bacterial 16S rRNA gene to show the constant and variable regions. Bottom: The two ITS (Internal Transcribed Spacer) regions located between the fungal 18S - 5.8S, and 5.8S - 28S rRNA genes (ETS: External Transcribed Space). The highly variable regions of the bacterial 16S and ITS rRNA gene regions are widely used in taxonomy and molecular phylogeny studies. (Figure recreated by Ashley Limon.)
Microbiome Sequencing
Sequencing Library Prep Pricing
For targeted amplicon sequencing, hypervariable regions spanning the entire bacterial 16S rRNA gene are covered by six amplicons. In the meantime, the ITS hypervariable region is also covered to uncover the diversity of fungal species. The library prep is provided at $70/sample from extracted DNA.
Library prep for metagenomic sequencing may use the TruSeq DNA or Nextera XT protocol depending on sample nature. Their rates are listed as Whole or Small Genome Sequencing Library Prep on the pricing page.
Metatranscriptomic sequencing library prep uses a process that is similar to total RNA-seq (i.e., employing bacterial rRNA depletion). If host rRNA and mRNA species are highly abundant, depletion of such host RNA molecules may be needed, which will incur additional charge (please inquire).
Sequencing
Targeted 16S and ITS rRNA gene sequencing is typically performed on MiSeq with read length up to 2x300 bp (PE300). Metagenomic and metatranscriptomic analysis typically require 2x150 bp (PE150) reads generated from HiSeq 4000 and NextSeq 500. As sequencing depth varies with project, please contact the core on the total number of reads needed.
Service Request
Project consultation is provided free-of-charge. ATAC-seq services can be requested through NUcore. Please finish and submit our project intake form when requesting this service.
Sample Requirement
Targeted 16S/ITS amplicon sequencing |
25 – 100 ng genomic DNA |
Metagenomic sequencing |
500 ng – 1 μg genomic DNA |
Metatranscriptomic sequencing |
300 ng – 3 μg total RNA |
Bioinformatics
Available upon request.
Frequently Asked Questions
What method should I use for DNA extraction?
The DNeasy PowerSoil Kit from Qiagen is a good general kit to try first. It’s always a good idea to conduct a literature search for what methods were used previously on similar sample types.
Is there a volume requirement for sample submission?
There is no specific requirement on sample volume. At least a few microliters of DNA/RNA samples is needed for processing. The total amount of DNA/RNA, as specified above, is more important than volume.
What if the total amount of DNA or RNA samples is lower than the required amount?
We might still be able to work with smaller amount. But to make sure we can get quality data from the samples, we’d suggest to submit a few trial samples first.
Can 16S and ITS rRNA be analyzed simultaneously to detect both bacterial and fungal groups?
Yes, NUSeq provides simultaneous analysis of both the entire bacterial 16S and fungal ITS rRNA genes.