Spatial Transcriptomics
Spatial transcriptomics enables researchers to study gene expression within the context of tissue architecture, microenvironments, and cell groupings—especially when paired with single-cell sequencing. To meet the growing demand for spatial -omics studies, NUSeq has joined forces with the Pathology Core Facility (PCF), the Mouse Histology and Phenotyping Laboratory (MHPL), and the Center for Advanced Microscopy (CAM) to form the Northwestern Spatial Consortium. This consortium provides investigators with access to the complete spatial analysis workflow: tissue preparation, cryosectioning, staining, imaging, tissue QC, sequencing library prep, sequencing, and data analysis. The workflow supports both fresh frozen (FF) and formalin-fixed paraffin-embedded (FFPE) tissues. Establishment of this comprehensive spatial pipeline is supported by the Northwestern University Office for Research.
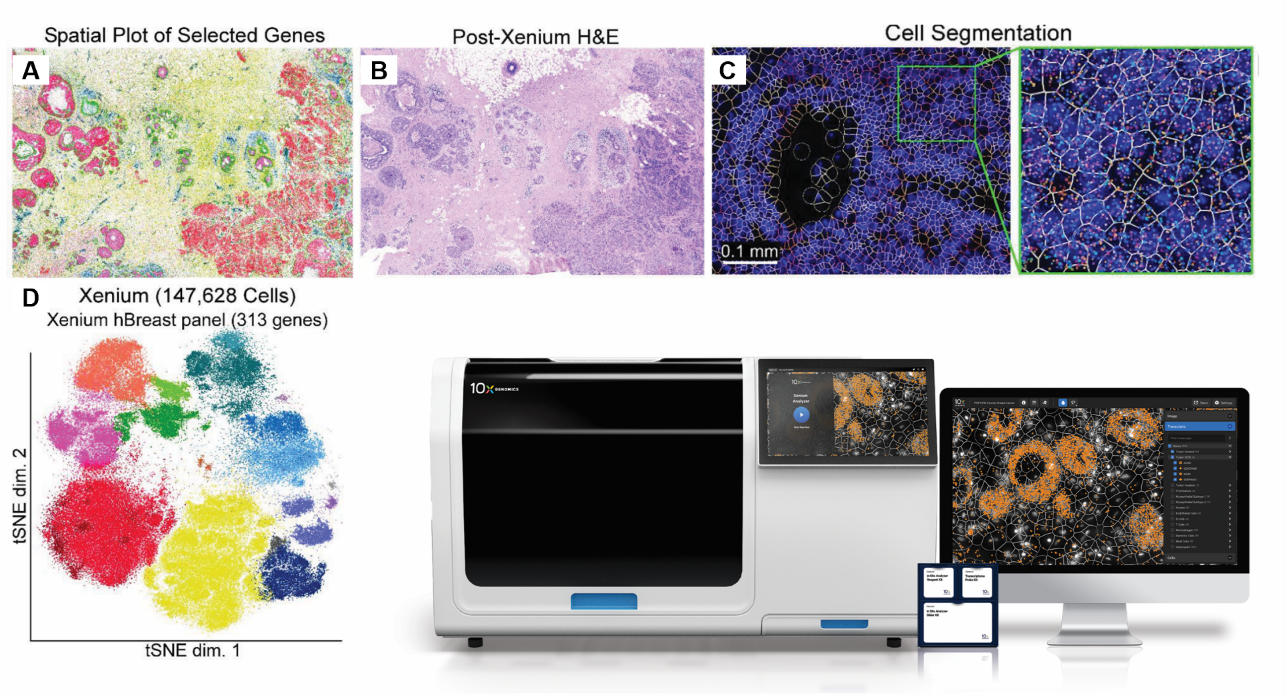
Xenium data from human breast cancer FFPE tissue using a 313-plex gene panel. A - Expression of selected gene markers (POSTN, IL7R, ITGAX, ACTA2, KRT15, VWF, CEACAM6, and FASN); B - H&E staining post Xenium workflow; C - Cell segmentation assigns transcripts to cells; D - t-SNE plot of different cell types in the Xenium data. (Panels A-D adapted from doi: https://doi.org/10.1101/2022.10.06.510405; Xenium image on the bottom right from 10x Genomics.)
Available Platforms
Xenium (10x Genomics)
- Technology: In situ hybridization-based targeted gene expression analysis (custom or pre-designed panels, up to 5,000 genes)
- Resolution: 200 nm optical, sub-50 nm localization with software
- Capture Area: 10.45 × 22.45 mm
- Sample Types: FFPE and fresh frozen
- Highlights: Supports IF, H&E staining, or Visium/Visium HD whole transcriptome analysis on the same section; integrates with 10x single-cell and Visium/Visium HD data
- Service Status: Available since September 2023; NUSeq has been a Certified Service Provider since November 2023
Visium/Visium HD (10x Genomics)

- Technology: Sequencing-based whole transcriptome analysis
- Resolution: 2 µm
- Capture Area: 6.5 × 6.5 mm (two capture areas per slide); larger 11 × 11 mm format upcoming
- Sample Types: FFPE and fresh frozen; pre-cut tissue sections on standard glass slides can be used; the Core’s 10x CytAssist instrument enables sample transfer from existing slides to Visium/Visium HD slides
- Highlights: Integrates spatial gene expression with brightfield and fluorescence microscopy images
- Service Status: Available since February 2022; NUSeq has been a Certified Service Provider since May 2022
Stereo-seq (STOmics)
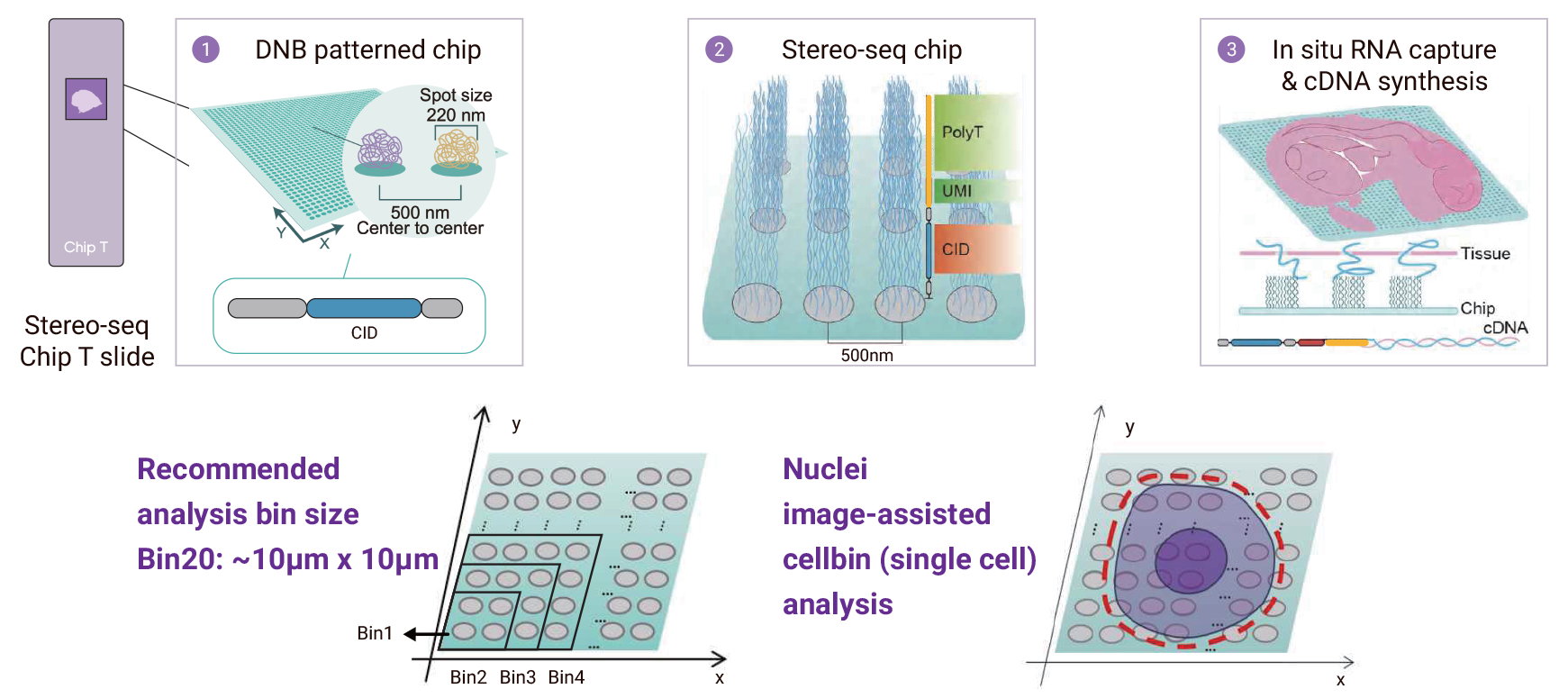
The Stereo-seq workflow.
- Technology: Sequencing-based whole transcriptome analysis
- Resolution: Spot size 0.22 µm, pitch 0.5 µm (subcellular resolution)
- Capture Area: Flexible, from 5 × 5 mm up to 13 × 13 cm custom chips
- Sample Types: FFPE and fresh frozen
- Highlights: Provides ultra-high resolution spatial maps across large, scalable tissue areas
- Service Status: Upcoming fiscal year 2026
Pricing by Platform and Stage
Xenium (10x Genomics)
- Slide Processing and Xenium Analyzer Run (up to 2 days): $4,900 for 2 slides
- Xenium Analyzer Run: $1,000 per day if additional day(s) needed
- Please check the Core Pricing webpage for external rates.
Visium/Visium HD (10x Genomics)
- CytAssist Visium v2 Slide Processing: $1,400 per 4 capture areas
- Visium HD Slide Processing: $1,600 per 4 capture areas
- Visium HD 3' Slide Processing: $1,800 per 4 capture areas
Stereo-seq (STOmics)
- Tissue Optimization on Test Slide: $420 per slide
- Spatial Gene Expression Library Prep: $980 per slide
- Please check the Core Pricing webpage for external rates.
Spatial Transcriptomics
Service Request
Please contact NUSeq to initiate a spatial analysis project. The Core works closely with rest of the Northwestern Spatial Consortium on the different steps of the workflow. Project consultation is provided free of charge. Consultation with the core prior to starting a spatial transcriptome experiment is highly recommended to ensure accomplishment of project goals.
Spatial transcriptomics services can be requested through NUcore.
Sample Submission
Sample Quality Control (QC)
All samples must pass QC before being accepted for a spatial transcriptomics service. To perform QC, core staff extracts RNA from the submitted material, and RNA quality is assessed using Bioanalyzer or TapeStation.
Tissue Compatibility
Xenium (10x Genomics)
- Fresh frozen tissue (embedded in OCT)
- Formalin-fixed paraffin-embedded (FFPE) tissue blocks
Visium/Visium HD (10x Genomics)
- Fresh frozen tissue (embedded in OCT)
- Formalin-fixed paraffin-embedded (FFPE) tissue blocks
- Fixed frozen tissue
- FFPE, fresh frozen, or fixed frozen sections mounted on regular microscope slides
- Archived Hematoxylin & Eosin (H&E)-stained FFPE slides
For detailed tissue preparation instructions, please see the latest demonstrated protocols for Visium/Visium HD and Xenium from the 10x Genomics site.
Stereo-Seq (STOmics)
- Fresh frozen tissue (embedded in OCT)
-
Formalin-fixed paraffin-embedded (FFPE) tissue blocks
For detailed preparation guides, please download the latest protocols for fresh frozen tissue and FFPE samples from the STOmics site. A list of validated tissues for the STOmics workflow is also available there.
Bioinformatics
Data analysis is provided upon request.
Frequently Asked Questions
Xenium (10x Genomics)
How do I start a Xenium experiment?
Please contact NUSeq (NUSeq@northwestern.edu) and we will help you through the whole workflow including sample preparation, probe designation, slide processing and data analysis.
Can I use fresh frozen or FFPE samples?
Yes, both are compatible with Xenium platform.
Can I use fixed tissue embedded in OCT?
How many samples can I fit on a Xenium slide?
The imageable sample area of Xenium slide is 10.45 mm x 22.45 mm and we have to run 2 slides per run. As long as the tissue can fit into the sample area, you can place as many sample as you want.
Can I study large tissue sections on Xenium?
Can different tissue types be placed on the same Xenium side?
Can the same slides be stained with H&E or other methods for imaging post a Xenium run?
Visium (10x Genomics)
What is the turnaround time?
The bottleneck is typically on identifying tissues with good RNA quality. Once sample(s) pass QC and section(s) are placed on Visium slide(s), the slide(s) will be processed in 2-3 weeks and the sequencing will be performed in two weeks.
How many tissue sections can each Visium Gene Expression Slide accommodate?
For Visium slide V1 with each capture area at 6.5mm x 6.5mm, four sections can be placed on each slide. For Visium slide V2 with each capture area at 11mm x 11mm, two sections can be placed on each slide.
Can I use tissue sections that have already been prepared on regular glass slides?
Yes. The core uses the 10x CytAssist instrument to transfer tissue analytes from existing slides to Visium slides for spatial analysis.
What is the spatial resolution of the 10x Genomics Visium spatial platform?
Each spot is 55 um in diameter, providing an average resolution of 1 to 10 cells per spot. The distance from center to center between spots is 100 um. Each capture area contains approximately either 5,000 (V1) or 14,000 (V2) barcoded spots.
What is the required RNA quality for an FFPE sample to proceed to spatial transcriptomic analysis?
DV200 score needs to be >30%. DV200, i.e., the percentage of fragments >200 nucleotides, is generated from Bioanalyzer or TapeStation traces.
How does the gene expression profiling process differ between fresh frozen and FFPE tissue samples?
For fresh frozen tissues which contain mostly intact RNAs, whole transcriptomic analysis is performed through poly-T-based cDNA reverse transcription followed by sequencing. Alternatively, probe-based hybridization method can be applied as well depends on project. For FFPE samples, due to presence of RNA degradation, this is achieved through hybridization of gene-specific probes followed by sequencing.
How many genes can be detected from fresh frozen or FFPE tissue sections?
18,000 human and 21,000 mouse genes.
What is the molecular capture rate, or sensitivity, of Visium?
Molecular capture efficiency is affected by a number of factors, including tissue type, permeabilization condition, and sequencing depth. Based on publicly available Visium data from 10x Genomics, the mouse brain hippocampus region as an example can generate an average of 4,500 genes and 20,000 unique transcripts per spot, at a tissue thickness of 10 um and sequencing depth of 50,000 paired-end reads per spot.
How to calculate the number of reads needed for my samples?
The suggested sequencing depth is 50,000 and 20,000 read pairs per spot at a minimum for poly-A capture and probe-based method, respectively. The number of spots needed to cover can be estimated from the percentage of capture area covered by the tissue section. For example, if 50% of the capture area is covered based on H&E staining image, the number of read pairs needed is: 50% coverage x 5,000 total spots per capture area x 50,000 read pairs per spot = 125 million read pairs.
What sequencing length is used for Visium libraries?
Read 1: 28 cycles; Index 1 (i7): 10; Index 2 (i5): 10; Read 2: 90.
Can I integrate spatial transcriptomics data with single-cell RNA-seq data from neighboring sections?
Yes. This is achievable. Please consult the core for more specifics.
How do I start a Visium experiment?
Please contact NUSeq (NUSeq@northwestern.edu) and we will help you through the whole workflow from sample preparation to Visium slide processing to data analysis.
Stereo-seq (STOmics)
What is Stereo-seq?
Stereo-seq (short for SpaTial Enhanced REsolution Omics-sequencing) is a next-generation spatial transcriptomics technology developed by STOmics. It’s designed to map the location of gene expression in tissues with ultra-high resolution and large field-of-view.
How does Stereo-seq work?
Stereo-seq uses DNA nanoball (DNB) arrays fixed on a patterned chip. Each DNB is labeled with a unique spatial barcode (known as a Coordinate ID, or CID) that corresponds to its precise location on the chip.
-
A tissue section is placed on the chip.
-
RNA transcripts released from the tissue are captured by DNBs on the chip and reverse-transcribed into cDNA, which then carries the spatial barcode.
-
The cDNA is made into sequencing libraries and sequenced.
-
Bioinformatics tools then map the sequencing data back to the original spatial coordinates, creating a detailed map of gene expression across the entire tissue section.
What is the size of the DNA nanoballs?
How many spots are there on a standard 1 cm x 1 cm chip?
A standard 1 cm x 1 cm Stereo-seq chip has 400 million spots.
What sequencers can I use to sequence Stereo-seq sequencing libraries?
How many reads do I need?
The recommended sequencing depth for the standard Stereo-seq 1 cm x 1 cm chip for fresh frozen samples is 1.5 billion reads (~4 reads per spot). For the Stereo-seq OMNI 1 cm x 1 cm chip for FFPE samples, the recommended sequencing depth is 4 billion reads (~10 reads per spot).